
“近年来,相继产生的疾病批改治疗药物扭转了脊髓性肌萎缩症(SMA)的人造病程,假设能在产生症状前尽早干预,患者会有很好的预后。”中国出世毛病干预救助基金会神经与肌肉疾病防控专项基金主任委员吴士文传授接受人民日报肥壮客户端采访时指出,倡导推进SMA的重生儿筛查,让SMA患儿尽早被发现。

对SMA展开重生儿以及基因携带筛查,一些兴旺国度及地域早已开局执行。
吴士文传授引见,2018年7月,美国将SMA归入了重生儿筛查的介绍一致目录。截至2023年2月,凑近99%的美国重生儿在出世时都会接遭到SMA筛查,从而可以早发现、早治疗。
2020年8月31日,欧洲SMA重生儿筛查联盟成立,联盟宗旨为在2025年前,让一切欧洲国度都能引入SMA重生儿筛查。
在我国,台湾地域是展开SMA重生儿筛查较早的地域,早在2006年就推行了重生儿普筛。在一些经济兴旺的东部沿海地域,比如广东省、山东省济南市、济宁市,江苏省的局部地域也曾经开局了这项筛查。
“和国外相比,咱们目前还没有成功普筛,现有的筛查多带有钻研性质,只要少局部病人被迫去筛查。”吴士文传授示意,假设有一天,SMA能够被归入国度级的普筛,会让更多患者被早发现、早治疗,从而取得更好的预后。
“目前,我国很多中央都在预备展开SMA重生儿筛查,假设缺少一致的规范,就存在必定危险。拿出一个能实真实在指点筛查的打算,十分有必要性和紧迫性。”吴士文传授示意。
近日,《脊髓性肌萎缩症重生儿筛查专家共识(2023版)》宣布在《中华医学杂志》上,对该病的筛查、诊断、治疗、随访启动了系统引见。作为通讯作者之一,吴士文传授以为,“共识的宣布,让中国SMA患者迈入了具备里程碑的新时代,推进了SMA的症状前治疗,也促成、完善与健全了稀有病治理体系。”
作为儿童神经病学专家,浙江大学医学院隶属儿童医院神经外科主任医师、浙江省SMA诊疗专家组组长毛姗姗传授向人民日报肥壮客户端引见,“关于SMA,遗传学诊断为金规范,目前筛查打算重要是针对SMN1 基因7号外显子纯合缺失的检测。筛查可以从小范围、单核心开局试点,逐渐将成功阅历推行到全国。”
“SMA的治疗就是在与期间赛跑,除了始终探求新药和新治疗手腕之外,越早诊断,越早开局规范治疗,预后就越好。重生儿筛查的目标就是为了更早的治疗,不然筛查就失去了意义。”毛姗姗传授坦言。
目前,世界范围内已上市的SMA疾病批改治疗药物有诺西那生钠注射液、利司扑兰口服溶液用散和索伐瑞韦三种。其中,诺西那生钠注射液和利司扑兰口服溶液用散已在国际获批并归入我国医保。随着SMA患者用药“最后一公里”的买通,治疗可及性的极大提高也反向助力于以后SMA治疗格式的变动。
那么关于经过重生儿筛查后确诊的SMA患儿,目前临床上症状前治疗的成果如何呢?毛姗姗传授引见道:“诺西那生钠注射液治疗症状前SMA患儿的NURTURE钻研已于近期发布5年随访结果,钻研数据标明,症状前SMA患者经诺西那生钠注射液治疗5年后的静止配置简直与反常孩子相反。”关于这个数据,毛姗姗传授的描画是“振奋人心”。
与此同时,毛姗姗传授也在治理着4位症状前治疗的SMA患者,第一个孩子在2月龄不到开局诺西那生治疗,11个月就可以独走,如今16个月了,跑、跳、高低楼梯跟反常孩子没有任何区别。“这就是症状前治疗的意义。”毛姗姗传授引见道。
让更多患儿成功症状前治疗,还需放大对共识的遍及和推行,把SMA患者尽早找进去。吴士文传授指出,首先,医生要在自己的畛域里启动推行、宣教、遍及,须要重生儿筛查、神经外科,遗传学等各个畛域的专家携手推进;此外,还要建设起SMA筛诊治一体化体系,让重生患儿失掉及时治疗,且能成功疾病的全流程治理。
此外,媒体乃至全社会也要增强对SMA筛查的科普宣传,让群众了解到,为重生儿做这项筛查不只具备卫生经济学的价值,也或者援救更多家庭。
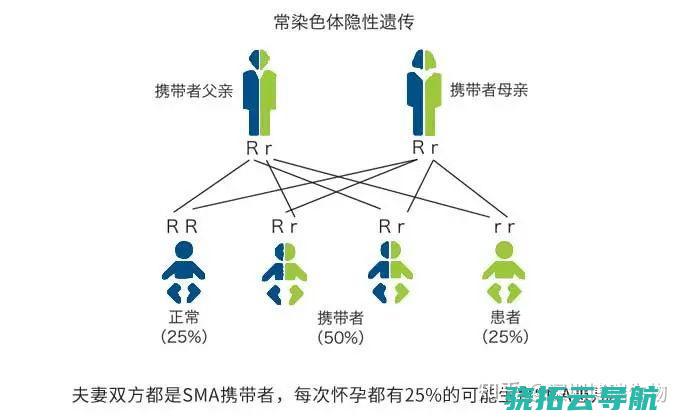
我们大家知道有一些新生儿在刚出生的时候因为体质是比较差的,所以容易患上一些疾病,而我们在这里便要来了解一下sma是什么症状?1300万一针“史上最贵药”获批临床?sma是什么症状 sma被称为小儿脊髓性肌萎缩症,医学上认为是身体的常染色体隐性遗传导致,也可能是基因突变引起病变,症状为四肢无力,身体的运动系统不控退化,但是sma携带者并不一定会发病。 sma又叫做脊髓型肌肉萎缩症,这种病是一种遗传性的先天性疾病。 因为sma是一种会导致肌肉无力和萎缩的运动神经元性疾病,患者会逐渐丧失各种身体功能,吞咽和呼吸功能也会受到影响。 一般来说,不累及面肌和眼外肌,不伴有腱反射亢进、不伴有感觉障碍。 肌肉的萎缩,随着年龄增长呈进行性加重的现象,肌电图是广泛的神经源性损伤,主要是累及前角的病变。 1300万一针“史上最贵药”获批临床 在新生儿中,SMA发病率为六千分之一至一万分之一,常规人群中,约每40人~50人就有1个是SMA致病基因携带者。 2018年5月,SMA被列入国家卫健委等部门联合制定的《第一批罕见病目录》。 与“渐冻人”同样残酷,SMA患者也会不断丧失对应的运动功能,只不过“渐冻人”一般是成年发病,而SMA一般是婴幼儿时期发病。 而且SMA是发病越早病情越重,“渐冻人”正好相反。 2019年5月24日,美国FDA批准了诺华公司的第一个用于治疗2岁以下SMA患者的基因疗法——Zolgensma。 患者只需接受一次静脉注射给药,就能在细胞中长期表达SMN蛋白,实现长期缓解甚至治愈。 sma携带者是什么意思 sma携带者,意思就是说身体有sma这种基因,这种病是一种遗传性的先天性疾病,症状为四肢无力,运动神经系统退化。 因为sma是一种会导致肌肉无力和萎缩的运动神经元性疾病,患者会逐渐丧失各种身体功能,吞咽和呼吸功能也会受到影响。 但是携带者不一定会发病,有点终身不会发作。 脊髓性肌萎缩症又称脊肌萎缩症,是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的疾病。 属常染色体隐性遗传病,临床并不少见。 根据发病年龄和肌无力严重程度,临床分为SMA-Ⅰ型、SMA-Ⅱ型、SMA-Ⅲ型,即婴儿型、中间型及少年型。 共同特点是脊髓前角细胞变性,临床表现为进行性、对称性、肢体近端为主的广泛性弛缓性麻痹与肌萎缩。 sma会遗传吗 会。 SMA是脊髓性肌萎缩症的英文缩写,是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的疾病,由多种基因突变引起,但一般特指由运动神经元存活基因1突变所导致,属常染色体隐性遗传病,父母都可以遗传给孩子。 SMA是常染色体隐性遗传病,夫妻双方可以进行DNA检测。 如果父母皆为携带者,则孩子有25%的几率是SMA;如果父母之中一方为携带者而另一方为SMA患者,则其后代有50%的几率是SMA;如果夫妻双方有一人不是SMA携带者,不论另一方是SMA患者还是携带者,孩子都不会成为SMA患者。
sma是什么病?SMA是脊髓性肌萎缩,S是脊髓,M是肌肉,A是萎缩,所以它的全称SMA是脊髓性肌萎缩。这是一种遗传性疾病,通常发生在婴儿期、儿童期和青春期。他的病理特征主要是脊髓前角细胞变性,主要临床表现为局限性进行性肌无力、肌萎缩、束颤。
一、大多数SMA1型最常见,其他为2型3型发生率最低。通过基因测试很容易识别这种疾病。目前发现一些运动神经元基因与之相关,因此有可能对一些基因进行诊断。为什么救命药能从70万降到3万多?3日上午,国家医疗保障局召开新闻发布会,公布2021年国家医保药品目录调整结果。被称为“高价药”的SMA治疗药物诺西辛钠注射液成功入榜。2019年4月28日,诺西酮钠注射液在国内上市,成为国内首个治疗SMA的药物。起初,69.97万针的报价让患者和家属刚刚燃起的希望瞬间熄灭。
二、多数病例会在一岁左右死亡,死亡原因多为心肌无力和呼吸衰竭。青少年发病患者病情进展缓慢,以下肢肌无力和萎缩最明显,股四头肌最早出现症状,肌无力由近端向远端发展。脊髓性肌营养不良是一种遗传性疾病,在sma早期表现为明显的症状,如四肢无力,运动神经系统变性,特别是吞咽困难,特别是呼吸功能,患者肌肉无力和萎缩。这种病在权威医院治疗效果比较好。sma是什么原因造成的?Sma是一种先天性疾病,主要由染色体隐性遗传引起。这种病会有家族使者,主要通过体外基因激活来治疗。
三、这种方法临床效果非常好,但是彻底治愈的希望还是很渺茫。但需要尽快接受系统治疗,恢复正常生活能力。一般的基因检测可以明确检测出这种疾病,这种疾病多发生在婴儿期和儿童期,也有发生在青春期,主要表现为肌肉萎缩或无力。
可以想像打一针就可以治疗好罕见的肌肉萎缩症,但却要花上新台币6700万元(212.5万美元)吗?
美国知名的诺华药厂日前获得美国食品和药物管理局(FDA)批准,上市一款治疗「脊髓性肌肉萎缩症」(SMA)的基因疗法 Zolgen *** a,适用于2岁以下的儿童病患。这种新药是世界上首创一次性治疗脊髓性肌萎缩症的药物,只要打一针就完成治疗。
而这样的罕病新药的价格为新台币6700万元(212.5万美元),也再次创下全球最贵药物的新纪录,打破了先前一款同样是基因疗法的眼科基因治疗药物Luxturna的纪录,这种药一剂定价为新台币2109万元(85万美元)。
所谓的「脊髓型肌萎缩症」(SMA),是因运动神经元基因突变所引起的罕见遗传性疾病,该基因存在于运动神经元蛋白中,这种功能性蛋白对运动神经细胞至关重要,而运动神经细胞则控制着全身的肌肉运动,如果缺少功能性蛋白会发生肌肉无力的症状,出现抬头、吞咽、呼吸障碍等问题。大多数患病幼儿,都因呼吸衰竭而无法幸免,会在2岁前死亡。
脊髓型肌萎缩症是一种遗传性疾病,是儿童发生率第二高的严重体染色体隐性遗传疾病。在台湾的 SMA 带原率约 2-4 %,大约每30到50人即有1人为带原者,若夫妻皆为 SMA 带因者,则每一胎儿无论男女皆有 1/4 的机会为 SMA 患者。新生儿发病率大约是 1/6000-1/,若以台湾每年有19万名新生儿计算,一年会有约10多个新生病例。
在Zolgen *** a研发上市之前,FDA在2016年9月核准一款Spinraza的药物来治疗脊髓型肌萎缩症,是第一个获准用于此疾病的药物,它是一种要注射到脊髓的反义寡核苷酸( ASO),总计10年的慢性治疗成本大约新台币1亿2750万元(400万美元),而且患者必须每4个月就要接受1次注射,需要终生注射,否则就会失效。而Zolgen *** a则是将新的DNA引进人体来纠正错误的基因,采取静脉注射,只要打一针就好,不必再像过去一样必须频繁进行脊髓穿刺注射。
而Zolgen *** a也不是完全没有副作用,副作用的表现为急性肝损伤带来的肝酶升高和呕吐,FDA 在批准要求中提到需要在标签中警示可能会患者会带来的副作用。
目前台湾食药署尚未核准这款新药进口,但国内多家大型教学中心型的医院都已经开始支持推动脊髓型肌萎缩症的基因筛检,以避免因遗传而带给下一代这种花费昂贵的罕见疾病。
相关标签: 助力SMA症状前治疗协助患者成功最大获益、 脊髓性肌萎缩症重生儿筛查、
本文地址:https://www.xiaotuo.net/hlwzxwz/3b198c14417022759ae6.html

近年来,相继产生的疾病批改治疗药物扭转了脊髓性肌萎缩症,SMA,的人造病程,假设能在产生症状前尽早干预,患者会有很好的预后,中国出世毛病干预救助基金会神经与肌肉疾病防控专项基金主任委员吴士文传授接受人民日报肥壮客户端采访时指出,倡导推进SMA的重生儿筛查,让SMA患儿尽早被发现,美国99%的重生儿,都启动S筛查对SMA展开重生儿以...。
明亮物流是提倡优质服务理念的专线物流公司,专注于中港物流领域,以南京物流、荔湾物流、越秀物流、海珠物流、天河物流、白云物流、黄埔物流、番禺物流、花都物流、南沙物流、从化物流、增城物流为核心,致力于为客户提供优质高效的货物运输服务。
云贝驾校网(www.ybjx.net)提供驾考2024科目一、科目三、科目四新题库模拟考试、c1仿真考试,帮助云贝考生迅速通过驾驶员理论考试!

吉林米莱格空间膜技术开发公司为您提供各种张拉膜、膜结构、空间膜的设计安装工程!竭诚服务,合作共赢!

温州网-温州市的网上新闻发布中心、公众信息服务中心和对外宣传的窗口。融新闻、全媒体、资讯、财经房产、时尚娱乐、旅游远足、教育培训、短信服务、互动直播、在线多媒体点播等各类信息和功能于一体。

爱收录网的宗旨是为中国网民提供更加优质的网络资源,更加便捷的上网体验,更加安全的网络环境。我们精心挑选各类优质网站,并不断更新和完善,让您畅享网络世界的乐趣。
何以笙箫成语网我们有生动的动画故事,有趣的成语谜语,成语对对子,看图猜成语还深刻的解释了成语出处,生动,有趣,好玩是我们的的建站思路,带给您完全不一样的成语大全网站。

东莞康耐德智能控制有限公司—连续5年高新技术企业,15年专注机器视觉系统、工业视觉检测、aoi检测,软硬一体化的研究与发展,在点胶AOI视觉检测行业的领域取得重大突破,为广大生产制造企业提供机器视觉一站式检测解决方案。

钉钉(DingTalk)是阿里巴巴集团打造的企业级智能移动办公平台,引领未来新一代工作方式,将陪伴每一个企业成长,是数字经济时代的企业组织协同办公和应用开发平台,是新生产力工具。
天下歌词搜集全天下所有歌词,提供LRC歌词下载,歌词找歌曲,歌词搜索等服务,网站歌词精心校队。是您搜索歌词的第一选择。